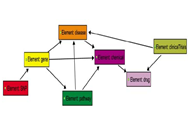
Single-nucleotide polymorphisms (SNPs) have been emerging out of the efforts to research human diseases and ethnic disparities. A semantic network is needed for in-depth understanding of the impacts of SNPs, because phenotypes are modulated by complex networks, including biochemical and physiological pathways. We identified ethnicity-specific SNPs by eliminating overlapped SNPs from HapMap samples, and the ethnicity-specific SNPs were mapped to the UCSC RefGene lists. Ethnicity-specific genes were identified as follows: 22 genes in the USA (CEU) individuals, 25 genes in the Japanese (JPT) individuals, and 332 genes in the African (YRI) individuals. To analyze the biologically functional implications for ethnicity-specific SNPs, we focused on constructing a semantic network model. Entities for the network represented by Gene, Pathway, Disease, Chemical, Drug, ClinicalTrials, SNP, and relationships between entity-entity were obtained through curation. Our semantic modeling for ethnicity-specific SNPs showed interesting results in the three categories, including three diseases (AIDS-associated nephropathy, Hypertension, and Pelvic infection), one drug (Methylphenidate), and five pathways (Hemostasis, Systemic lupus erythematosus, Prostate cancer, Hepatitis C virus, and Rheumatoid arthritis). We found ethnicity-specific genes using the semantic modeling, and the majority of our findings was consistent with the previous studies that an understanding of genetic variability explained ethnicity-specific disparities.
Single-nucleotide polymorphisms (SNPs) have been emerging out of the efforts to research human diseases and ethnic disparities. A semantic network is needed for in-depth understanding of the impacts of SNPs, because phenotypes are modulated by complex networks, including biochemical and physiological pathways. We identified ethnicity-specific SNPs by elim...
Single-nucleotide polymorphisms (SNPs) have been emerging out of the efforts to research human diseases and ethnic disparities. A semantic network is needed for in-depth understanding of the impacts of SNPs, because phenotypes are modulated by complex networks, including biochemical and physiological pathways. We identified ethnicity-specific SNPs by eliminating overlapped SNPs from HapMap samples, and the ethnicity-specific SNPs were mapped to the UCSC RefGene lists. Ethnicity-specific genes were identified as follows: 22 genes in the USA (CEU) individuals, 25 genes in the Japanese (JPT) individuals, and 332 genes in the African (YRI) individuals. To analyze the biologically functional implications for ethnicity-specific SNPs, we focused on constructing a semantic network model. Entities for the network represented by Gene, Pathway, Disease, Chemical, Drug, ClinicalTrials, SNP, and relationships between entity-entity were obtained through curation. Our semantic modeling for ethnicity-specific SNPs showed interesting results in the three categories, including three diseases (AIDS-associated nephropathy, Hypertension, and Pelvic infection), one drug (Methylphenidate), and five pathways (Hemostasis, Systemic lupus erythematosus, Prostate cancer, Hepatitis C virus, and Rheumatoid arthritis). We found ethnicity-specific genes using the semantic modeling, and the majority of our findings was consistent with the previous studies that an understanding of genetic variability explained ethnicity-specific disparities.