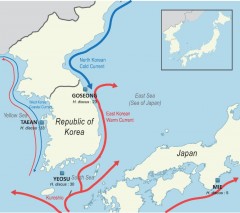
Continuous monitoring of the present genetic status is essential to preserve the genetic resource of wild populations. In this study, we sequenced regional Pacific abalone Haliotis discus samples from three different locations around the Korean peninsula to assess population structure, utilizing Genotyping-by-Sequencing (GBS) method. Using PstI enzyme for genome reduction, we demonstrated the resultant library represented the whole genome region with even spacing, and as a result 16,603 single nucleotide variants (SNVs) were produced. Genetic diversity and population structure were investigated using several methods, and a strong genetic heterogeneity was observed in the Korean abalone populations. Additionally, by comparison of the variant sets among population groups, we were able to discover 26 Korean abalone population-specific SNVs, potentially associated with phenotype differences. This is the first study demonstrating the feasibility of GBS for population genetic study on H. discus. Our results will provide valuable data for the genetic conservation and management of wild abalone populations in Korea and help future GBS studies on the marine mollusks.
Continuous monitoring of the present genetic status is essential to preserve the genetic resource of wild populations. In this study, we sequenced regional Pacific abalone Haliotis discus samples from three different locations around the Korean peninsula to assess population structure, utilizing Genotyping-by-Sequencing (GBS) method. Us...
Continuous monitoring of the present genetic status is essential to preserve the genetic resource of wild populations. In this study, we sequenced regional Pacific abalone Haliotis discus samples from three different locations around the Korean peninsula to assess population structure, utilizing Genotyping-by-Sequencing (GBS) method. Using PstI enzyme for genome reduction, we demonstrated the resultant library represented the whole genome region with even spacing, and as a result 16,603 single nucleotide variants (SNVs) were produced. Genetic diversity and population structure were investigated using several methods, and a strong genetic heterogeneity was observed in the Korean abalone populations. Additionally, by comparison of the variant sets among population groups, we were able to discover 26 Korean abalone population-specific SNVs, potentially associated with phenotype differences. This is the first study demonstrating the feasibility of GBS for population genetic study on H. discus. Our results will provide valuable data for the genetic conservation and management of wild abalone populations in Korea and help future GBS studies on the marine mollusks.